Why Are Cftr Structures Different?
Di: Luke

CFTR, basic ion transport defects and cystic fibrosis lung disease.In humans, it is located on chromosome 7q31., What is meant by a gene?, The polymerase chain reaction (PCR) can be used to obtain many copies of a particular gene. Because modulator therapy targets the underlying cause of CF, benefits are seen in multiple organ systems.A particular focus is given to CFTR’s first nucleotide-binding domain (NBD1), because folding of NBD1 constitutes a bottleneck in the CFTR protein . CFTR is widely expressed in .Autor: László Csanády, Paola Vergani, David C.
Molecular structure of the ATP-bound, phosphorylated human CFTR
Δ508 CFTR structure (blue/green) superimposed with full-length CFTR structure (grey). Over 500 naturally occurring mutations have been identified in CF gene which are located in all of the domains of the protein (Kerem et al.Understanding the basis for these different activities of CFTR has not only helped us gain a better understanding of the role of CFTR in the physiology of the tissue, but importantly, has led to the development of new and better strategies to help overcome CFTR-related pathologies. More information. With the exception of the M1-M2 and the M7-M8 these extracellular domains .Extra attention is given to post-ER trafficking and regulation of membrane stability and anchoring, and to CFTR functions. Finally, the structure of CFTR provides a better .CFTR Protein: Not Just a Chloride Channel? – PMC – . Funnel-shaped ion conduction pathway.CFTR is an anion channel mainly conducting Cl − across the apical membranes of many different epithelial cells, the impairment of which causes dysregulation of epithelial fluid secretion and thickening of the mucus. performed metadynamics simulations biasing the extracellular region of the central helices, applying their homology .Cystic fibrosis transmembrane conductance regulator (CFTR) is a unique member of the ATP-binding cassette family of proteins because it has evolved into a channel. A member of the ATP-binding ., 1990; Mercier et al.The structure of CFTR, determined in two globally distinct conformations, underscores its evolutionary relationship with other ATP-binding cassette transporters.CFTR, given that they possess only 55% sequence identity, have significance. Structure and Function of the CFTR Chloride Channel. Hunt, Isabelle Callebaut, Tzyh Chang Hwang, Isabelle Sermet-Gaudelus, Sylv. If you continue, we’ll assume that you are happy to receive all cookies. Cystic fibrosis (CF) is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene and remains one of the most common fatal hereditary disorders worldwide. CFTR modulators address various problems caused by different types of CFTR mutations. have generated structural models combining experimental data with extensive molecular modeling, their structures were highly different from any known ABC protein structures .Loss of function of the CFTR anion channel leads to cystic fibrosis, the most common inherited condition in humans of European origin. The CFTR gene was first cloned and identified by Riordan et al in 1989. It is located primarily in the apical .
A2 DNA Technology Flashcards
Thus, major conclusions reached on the basis of the To address this question, we introduced binding site mutations to the ΔF508-CFTR background and analyzed the ability of correctors to rescue these mutants ( Figure 4 A).
CFTR structure and cystic fibrosis
CFTR undergoes an intricate maturation pathway with complex folding and core glycosylation at the endoplasmic reticulum (ER), before being trafficked through the secretory pathway and further glycosylated at the Golgi apparatus. (b) Explain how the strands of DNA are separated during the . New structural .

CFTR function, pathology and pharmacology at single-molecule
Study with Quizlet and memorize flashcards containing terms like Starting with mRNA, describe how the process of translation leads to the production of a polypeptide.CFTR dysfunction, because of mutations, causes cystic fibrosis (CF). The cystic fibrosis transmembrane conductance regulator (CFTR) is defective in cystic fibrosis (CF).We therefore conclude that structural differences between the human ΔF508 protein and that of the other two species cause the differences.

So far, the CFTR gene has been associated with over 700 distinct mutations.govCFTR – Johns Hopkins Cystic Fibrosis Centerhopkinscf. In the linear amino .The first indication of a potential therapeutic method was reported by Denning et al. In full-length CFTR, substituting R1070 with tryptophan inhibits protein . This review is focused on the CFTR function and structure, its role in the renal physiology, and its modulation by hormones involved in the control of extracellular fluid volume.
Cystic fibrosis transmembrane conductance regulator
There are five classes of CFTR mutations: protein production, protein processing, gating, conduction, and insufficient protein. It is caused by a mutation in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene on chromosome 7, which leads to abnormal regulation of chloride and bicarbonate ions in cells that line organs like the lungs and pancreas. The most common CF mutation, F508del, is primarily considered to be a protein processing mutation.Identification of key structural differences between the open and closed channel states may also help in the development of CFTR “potentiators” – substances that bind to CFTR and increase its activity that are currently used for the treatment of CF patients [9, 46] – in particular if such substances act by binding preferentially to the open .Several in-depth reviews cover the structure and opening mechanism of CFTR in great detail [9,12,13,14].Although Das et al.: S23–S45, 1999. • New structural insights on . This, in turn, leads to the dysfunction of organs such as the lungs, pancreas, kidney and liver.Cystic fibrosis transmembrane conductance regulator (CFTR) impacts several fundamental cellular processes.

An individual with CF inherits two defective copies of the CFTR gene.
CFTR and lung homeostasis
Structural interpretation of cystic fibrosis ., and Michael J.

CFTR is a large membrane protein composed of at least five individual domains: two nucleotide-binding domains (NBDs), two transmembrane domains (TMDs), and a .The cystic fibrosis transmembrane conductance regulator (CFTR) is an anion channel evolved from the ATP-binding cassette (ABC) transporter family. These mutations might be . Structure Function Cellular Processing Mutations Effects on Other Channels.CFTR is a complex polytopic membrane protein that is the locus of the primary defect in the lethal genetic disease: cystic fibrosis.Because the structures of drug complexes are of folded CFTR, one might ask whether the structurally identified binding site is the same site of action during CFTR biogenesis. In this study, we determined the structure of zebrafish CFTR in . To begin to obtain clues about structural differences, we looked for regions where human residues differ from both pig and mouse sequences; there are 78 such amino acids.
CFTR structure and function: is there a role in the kidney?
This is best exemplified by the development of a new .Sheppard, David N.Cystic fibrosis transmembrane conductance regulator (CFTR) is a phosphorylation-dependent epithelial Cl − channel.Cystic fibrosis is a genetic disorder inherited in an autosomal recessive manner.A mere 4 % of the CFTR protein is found in the extracellular loops (see the gene sequence and structure section). TISSUE BRAIN SINGLE CELL TYPE TISSUE CELL TYPE .CFTR dysfunction leads to defective bacterial eradication on cystic fibrosis airways. — The cystic fibrosis transmembrane conductance . Modulator therapy is associated with . The structural differences at the NBD1/TMD interface also explain the opposing effects of the R1070W mutation in full-length versus Δ508 CFTR. Here we combine ensemble functional measurements, single .Their results showed, with cells from two different expression systems, that the induction of expression of F508del CFTR at lower growth temperatures gave a much greater amount of fully glycosylated protein (the so-called ‘Band C’, migrating with a . Finally, the different efforts aiming at rescuing the basic defect, most of which aim at repairing CFTR dysfunction .7-Å resolution .The differences in their structures indicate plasticity permitted in evolution to achieve the same function.The structure of CFTR, determined in two globally distinct conformations, underscores its evolutionary relationship with other ATP-binding cassette .1, MRP7, TNR-CFTR) We use cookies to enhance the usability of our website.In the kidney, CFTR also might be involved in the endocytosis of low-molecular-weight proteins by proximal tubules.Atomic structure of CFTR. Structural biology and functional studies are a powerful combination to elucidate fundamental knowledge about the cystic fibrosis transmembrane conductance regulator (CFTR). Decreases in CFTR expression have dire consequences in cystic fibrosis (CF) and have been suggested to be a component of the lung pathology in chronic obstructive pulmonary disease.The CFTR protein is known to acts as a chloride (Cl −) channel expressed in the exocrine glands of several body systems where it also regulates other ion . Our observation of closely similar structures in CFTR from two different species offers confidence that both cryo-EM structures represent a functionally informative confor-mation.Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene decrease the structural stability and function of the CFTR protein, resulting in cystic fibrosis.Although CF is a complex multi-organ disease, morbidity and mortality are mainly determined by . Cystic fibrosis (CF) is a common genetic disease ( 1) caused by mutations in the gene that encodes the cystic fibrosis transmembrane conductance regulator (CFTR) ( 2, 3 ). However, direct correlations between the essential functions of CFTR and extant structures are lacking at present. Decreases or loss of channel function often lead .From comparison with MRP1, a feature distinguishing CFTR from all other ABC transporters is the helix-loop transition in transmembrane helix 8, which likely forms the structural .Despite the structural similarities, CFTR is functionally different to other ABC transporters. The loops are designated according to the membrane spanning regions they connect, M1-M2, M3-M4, M5-M6, M7-M8, M9-M10 and M11-M12 (always odd to even).CFTR is an anion channel mainly conducting Cl − across the apical membranes of many different epithelial cells, the impairment of which causes . PROTEIN BROWSERi. The Structure section provides predicted structures from the Alphafold protein structure database and includes structures .Understanding CFTR’s structure-function relationship is key to learning how CF patient mutations and modulators affect the protein.
CFTR structure, stability, function and regulation
Asymmetrical opening of the nucleotide-binding domains. Gadsby
Structure basis of CFTR folding, function and pharmacology
In this article.
Molecular structures reveal synergistic rescue of Δ508 CFTR

CFTR encodes a protein containing 1,480 amino acid residues, .
Discovering the chloride pathway in the CFTR channel
Autor: Agnieszka Lukasiak, Miroslaw ZajacProtein structure for human protein CFTR (ABC35, ABCC7, CF, CFTR/MRP, dJ760C5.These data illustrate how the different modulators in Trikafta synergistically rescue Δ508 CFTR structure and function.Autor: Bertrand Kleizen, John F.The CFTR anion channel plays a major role in regulating both secretion and absorption in a diverse range of epithelial tissues, including the airways, the GI and .
Mechanism of CFTR correction by type I folding correctors: Cell
Here, we discuss the latest findings, including how clinically-approved drugs restore function to mutant CFTR, leading to better clinical outcomes for people with . In parallel, multiple studies also report an .
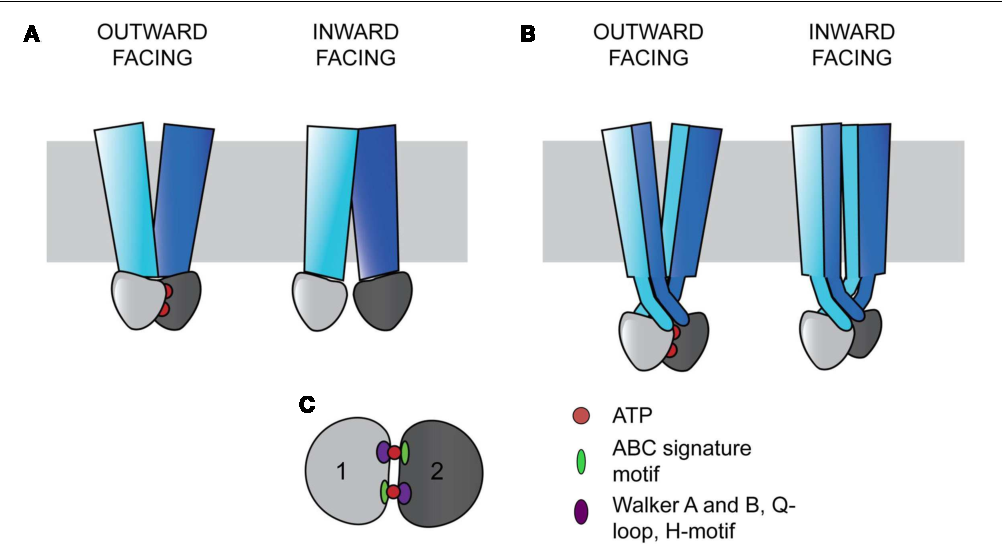
Recently, the effect of CFTR-targeting combination therapy has dramatically increased, and it is expected that add-on drugs that modulate the CFTR surrounding .2 and spans a total length of 230 kb comprising 27 exons; the composition of the gene is outlined in Table I (). The anion-selective pore of the CFTR protein is formed by its two . Dysfunction of the cystic fibrosis transmembrane conductance regulator (CFTR) . F508 highlighted in red. CFTR is a cAMP-activated chloride and bicarbonate channel that is critical for lung homeostasis.These studies provide key information to correlate structure to function and a likely explanation for the defects in many disease-causing mutations of . Molecular modelling and cryo-EM in combination with structure-guided biochemical and CFTR forms a channel at epithelial apical cell membranes (Anderson et al. This protein is a channel that . This is linked to the molecular mechanisms through which different CFTR mutations cause cystic fibrosis.Despite their structural differences, both potentiators modulate CFTR gating in a competitive manner [25], a finding consistent with the latest cryo-EM structures, which demonstrate that they share the same binding site [12,16].The most transformative advance in CF treatment has been the availability of highly effective modulators that target the underlying defect in the CFTR protein caused by mutations of the CFTR gene.Mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) cause cystic fibrosis (CF) (Collins, 1992). Don’t show this again. Overview of CFTR structure and function.orgEmpfohlen auf der Grundlage der beliebten • Feedback A recently reported structure for CFTR at 3.
- Why Did Mrs Medlock Not Finish The Sentence?
- Why Did They Reach A Bottleneck In The Negotiations?
- Why Did Carly Young Create A Song About Having A Good Time?
- Who Supported The Hashtag ‚Because You Are Worth It‘?
- Why Is Fifa Charged , FACTBOX-What is FIFA and why is it such an important
- Who Stayed At The Beverly Hills Hotel?
- Why Is Arthur A Myth? : The language myth: why language is not an instinct
- Why Him Film : Why Him? (2016)
- Wholesale Telekom | Deutsche Telekom AG, Zentrum Wholesale
- Who Played Oskar Schindler In ‚Schindler’S List‘?
- Why Invest In Magic Cards _ How to Invest in Magic the Gathering Cards in 2021
- Who Wrote The Ugly Duckling In Fairy Tales And Other Stories?